Challenge
Identification of Active Small Molecules for Drug Discovery challenge

What is this challenge all about?
Drug developers still rely heavily on experimental trial and error cycles to identify and optimize small molecules. Typically, well over 1000 molecules need to be synthesized and tested per program to identify a clinical candidate. Thus, developing a new clinical grade compound comes at a great cost of a range of $10s of millions and 3+ years.
To design and develop effective small molecules, the existing process requires an accurate 3D model of the target protein and a pocket in which a small molecule should bind. The next step is to screen a vast space to identify potential hits (discovery) followed by a refined mechanism to prioritize and rank hits by their affinity (hit ranking / optimization).
While we have seen progress in the field of computational small molecule design, there are significant limitations in each step. Virtual screens potentially work well when the target is accurately modeled and molecules that bind in the desired location are known, but docking approaches often fail to identify active molecules due to deficiencies in representing the physics of small molecule and protein interaction. Even when applying secondary filters such as empirical or physics-based scoring to rank order hits, calculations are still inaccurate, expensive, slow, and not suitable for rank ordering of diverse molecules.
The desired solution
The desired solution should be focused on an AI/Machine learning methodology beyond the traditional computational methods of hit discovery by virtual screening. We are looking for software tools that can be adopted by pharma companies and will improve accuracy and speed in identifying active molecules for drug discovery. These tools should be based on learning approaches rather than brute force.
We envision the following objectives which can be executed as a single sequential workflow or individually:
1. Modelling the target for small molecule design A target protein without an experimentally defined structure or pocket and no high homology structures to other protein is available (protein sequence identity < 40%). We may have some idea where we want the molecule to bind the target but the models are inaccurate.
2. Hit discovery Starting with a proprietary library of millions of compounds and computationally identifying dozens of high affinity hits. No known binding molecules will be provided and candidates will be validated using biochemical, cell activity, or functional assays and not necessarily binding affinity assays. Predicting off targets, toxicities, permeability and other ADME properties is out of scope.
3. Ranking the hits for binding affinity Accurately predict the binding affinity of hundredsof synthesizable compounds under consideration by medicinal chemists. This objective requires high accuracy and wider scope of chemical diversity.
Briely, the input to the platform will be the target sequence and an optional small molecule library. The output will be a ranked ordered list of dozens of molecules by their binding affinity.
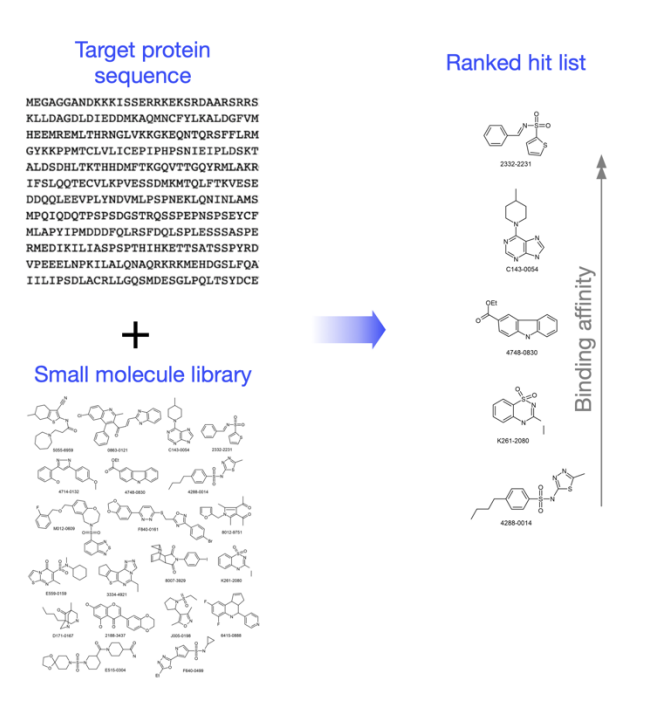
How will you measure success?
The new technology developed by the company will be tested on ongoing small molecule discovery projects or on specific targets of interests provided by the pharma partners. The proof of conecept will focus on finding and ranking the hits as a first step. Initially, testing will be limited to available compounds that do not require a new synthesis.
Will pharma companies provide data from small library screens?
Yes. For a well defined need, specific data can be made available upon request.
Can you provide an example that will demonstrate the impact of this technology and the complexity to address?
To understand the potential impact, we will first address the existing approach which typically follows these steps:
• Starting with a known active molecule, medicinal chemists or computational chemists build hypotheses of molecular features to keep or modify or improve activity, then synthesize and test the molecules.
• The “known active” molecules could come from experimental data or literature molecules. The “quality” of hypotheses is based on individual’s prior knowledge and current data of the molecules.
• The cycle time of this process is from weeks to months before we know if the molecule achieves the desired potency, then the cycle of design-test-make continues.
• If there is no “known active” molecule, experimental high-throughput screening (HTS) or virtual screening (VS, described above) will be used to identify active molecules. HTS requires significant resources in assay development, availability of material molecules, and time to run the experiment. VS could be done computationally, but success rate is low. The VS’s binding affinity prediction (or ranking ordering) often does not correlate with experimental activity measurements.
The ability to computationally predict high quality molecules for a common scenario where the target protein is unknown, the library of small molecules is provided and there are no known ligands as a starting point – would save extensive costs of experimental high throughput screening and binding site mapping. In addition, the ability to rank order the hits effectively will reduce signicantly the experimental validation time and cost.